一、背景介绍
锂硫(Li-S)电池因其高度理论容量(2600Whkg-1)和硫的环保特性而有望成为下一代具有高能量密度的储能设备。然而,硫的氧化还原反应在动力学上非常缓慢。因此,加速硫氧化还原动力学是实现高性能锂硫电池的必要条件。将电催化剂引入硫正极能够解决上述动力学问题。例如,具有高价金属元素的金属二硫化物[二硫化钼(MoS2)]表现出强的路易斯酸性,因此在Li-S电池中能够增加硫氧化还原动力学。然而,电解质中具有路易斯碱性位点的锂盐和溶剂也不可避免地与电催化剂上的路易斯酸性位点相互作用。因此,有必要阐明电催化剂与锂盐以及溶剂之间的相互作用,以深入了解电催化硫氧化还原反应的工作机理,合理设计电催化剂和电解质以构建高性能的Li-S电池。
二、正文部分
01成果简介
近日,清华大学张强教授联合北京理工大学李博权副研究员,通过研究发现双(三氟甲磺酰基)亚胺锂(LiTFSI)会加剧MoS2电催化剂的表面凝胶化。LiTFSI中的三氟甲磺酰基与MoS2电催化剂上的路易斯酸性位点相互作用,产生一个缺电子中心。具有高路易斯酸度的缺电子中心引发1,3-二氧戊环溶剂的阳离子聚合并产生降低电催化活性的表面凝胶层。为了解决上述问题,他们通过引入路易斯碱碘化锂(LiI)来阻断LiTFSI与MoS2之间的相互作用并抑制表面凝胶化。因此,采用MoS2电催化剂和LiI添加剂的Li-S软包电池实现了416Wh kg-1的超高能量密度。该研究以题目为“RegulatingLithium Salt to Inhibit Surface Gelation on an Electrocatalyst forHigh-Energy-Density Lithium–Sulfur Batteries”的论文发表在国际顶级期刊《Journalof the American Chemical Society》上。

02研究亮点
本工作证明LiTFSI会在Li-S电池中加剧MoS2电催化剂的表面凝胶化。为了解决上述问题,本工作将路易斯碱盐碘化锂(LiI)引入到Li-S电池中,以阻断LiTFSI与MoS2之间的相互作用,避免产生缺电子位点,并抑制后续表面凝胶化。因此,具有LiI添加剂和MoS2电催化剂的Li-S电池在表现出更高的倍率性能、高的放电容量和416Wh kg–1的超高实际能量密度。
03图文导读
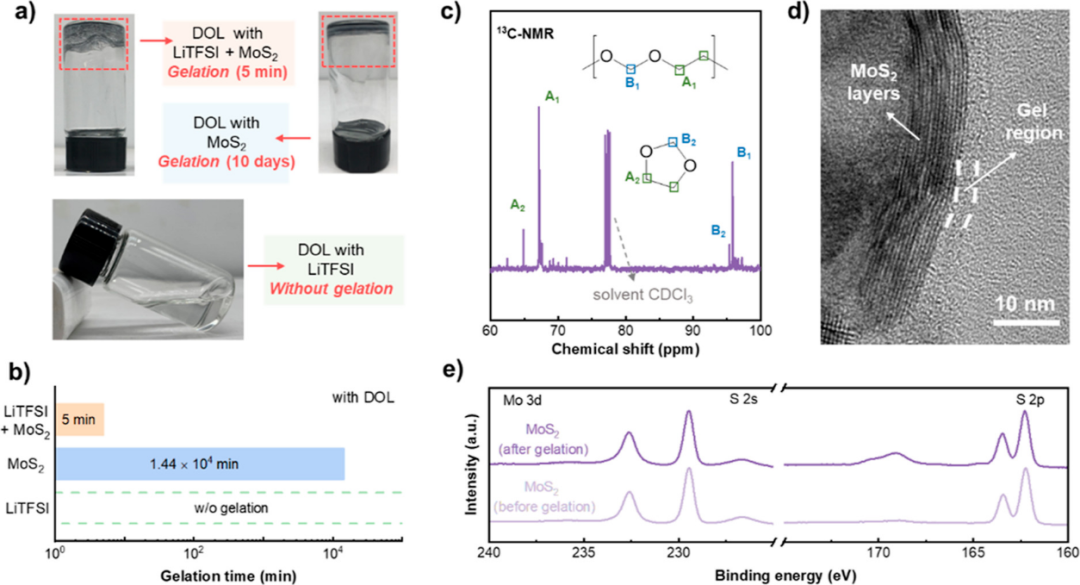
【图1】(a)凝胶现象的光学图像和(b)DOL与LiTFSI+MoS2、MoS2或LiTFSI混合时的凝胶化时间。(c)凝胶的13CNMR光谱。(d)凝胶化过程后MoS2电催化剂的TEM图像。(e)表面凝胶前后MoS2电催化剂的高分辨率XPSMo 3d和S2p光谱。
图1a显示,将MoS2、LiTFSI和DOL混合在一起时,准固体凝胶迅速生成,MoS2颗粒紧紧地粘在瓶底,表明MoS2在凝胶化中起重要作用。此外,DOL、LiTFSI和MoS2系统的凝胶化过程仅在5分钟内快速完成,但DOL和MoS2系统的凝胶化过程耗时约10天(图1b),表明凝胶化速率取决于LiTFSI的添加。简而言之,MoS2触发了凝胶化,而LiTFSI加剧了凝胶化,表明MoS2电催化剂、DOL溶剂和LiTFSI盐之间存在复杂的相互作用。
13C核磁共振(13CNMR)谱显示,出现在67.1ppm(A1)和95.8ppm(B1)的信号分别代表-O-CH2-CH2-O-和-O-CH2-O-中的碳原子(图1c)。因此,得到的凝胶是DOL溶剂的开环聚合产物。图1d的TEM图像显示,纳米尺寸的凝胶层紧紧包裹着MoS2纳米颗粒。同时,X射线光电子能谱(XPS)结果显示,MoS2电催化剂在凝胶化前后的Mo和SXPS峰具有相同的结合能(图1e)。因此,凝胶化不会影响整体性能,但会在MoS2电催化剂表面生成凝胶层,这会影响Li-S电池的电催化性能。
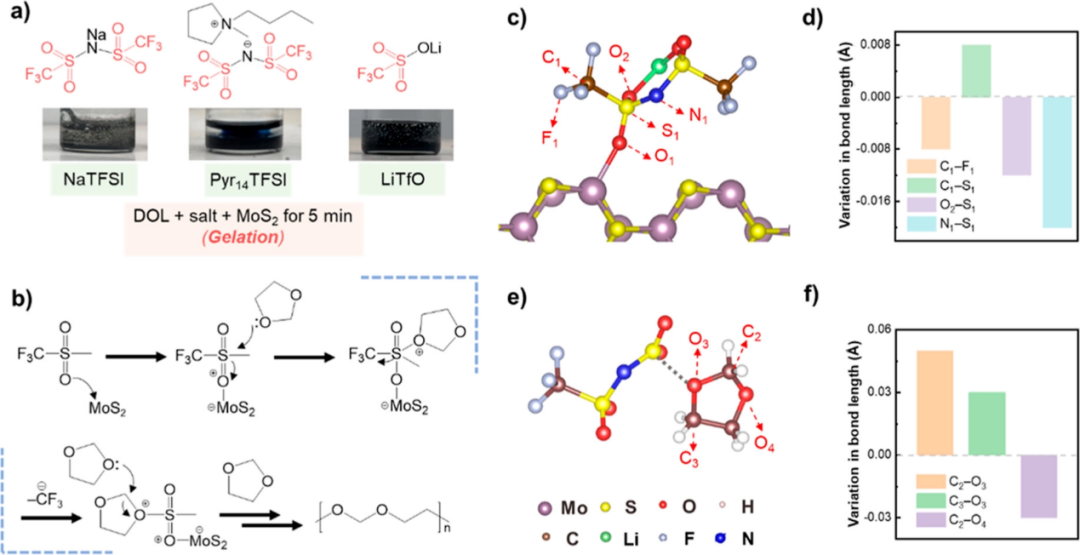
【图2】(a)DOL+盐+MoS2系统(盐=NaTFSI、Pyr14TFSI或LiTfO)中凝胶现象的光学图像。(b)CF3SO2-基团加速DOL在MoS2电催化剂上开环聚合的反应机理图。(c)LiTFSI分子与MoS2(110)表面相互作用的侧视图和(d)在MoS2(110)表面吸附前后键长的变化。(e)DOL分子与LiTFSI剩余部分相互作用的俯视图和(f)相互作用前后键长的变化。
首先研究了Li+是否是凝胶加剧的根源。图2a显示,通过使用NaTFSI或Pyr14TFSI,将LiTFSI中的Li+转变为Na+或具有超弱路易斯酸性的有机阳离子1-丁基-1-甲基吡咯烷(Pyr14+)(图2a)。然而,凝胶化仍然迅速发生,表明加剧的表面凝胶化与Li+无关。接下来,检查了带有Lewis碱性N和O原子的TFSI-,它们可以与Lewis酸性MoS2电催化剂相互作用,并影响表面凝胶化过程。为此,N原子被O原子取代,以检查N原子是否是与MoS2相互作用并触发凝胶化的实际位点。采用三氟甲磺酸锂(LiTfO)替代LiTFSI,凝胶化过程也在5分钟内完成,类似于LiTFSI的凝胶化过程(图2a),表明路易斯碱性N原子不是活性位点。因此,CF3SO2-基团中的O原子可能与MoS2相互作用并极大地激活随后的凝胶化。为了验证上述论点,将三氟甲磺酸甲酯(TfOMe),一种仅含有CF3SO2–的非电离分子与DOL和MoS2混合。结果显示,流体迅速变成凝胶相,类似于LiTFSI。因此,LiTFSI中用于激活凝胶化过程的官能团是CF3SO2-基团中的路易斯碱性O原子,而不是Li+或N原子。
为了解释CF3SO2–基团中Lewis碱性O原子在加剧表面凝胶化中的具体作用,基于Lewis酸碱理论提出了一种DOL开环聚合链引发机制(图2b)。具体而言,CF3SO2–基团中的O原子与MoS2电催化剂中的路易斯酸性Mo原子相互作用,O2p的孤对电子与Mo3d空轨道共享形成氧鎓离子。然后,正电中心转移到CF3SO2–基团中相邻的S原子上,产生一个缺电子S中心,其路易斯酸性比MoS2电催化剂中Mo原子的原始路易斯活性位点更强。随后,DOL溶剂中O原子的孤对电子攻击Lewis酸性S原子并引发CF3-基团的损失。随后,在DOL分子的O原子上产生一个正电中心,并引发随后的DOL开环聚合。考虑到开环聚合的速率决定步骤是DOL的活化,活性位点的路易斯酸度越高,表面凝胶化的速率越高。LiTFSI和MoS2电催化剂之间的相互作用通过Mo-O-S三原子相互作用提供了一个高活性的缺电子S中心,其路易斯酸度高于MoS2电催化剂中的Mo原子。因此,表观凝胶速率是由LiTFSI和其他具有CF3SO2-基团的分子决定的。
为了验证上述假设机制,进行了密度泛函理论(DFT)计算。首先,当LiTFSI分子吸附在MoS2表面时,CF3SO2-基团中的O原子通过Mo-O1键以2.16Å的键长与MoS2(110)结合(图2c)。同时,LiTFSI内的电荷密度发生了变化,导致相应的键被削弱或加强。键长分析显示,C1-S1键长增加了0.008埃,而其他主要化学键包括C1-F1、O2-S1和N1-S1键分别缩短了0.008、0.012和0.020埃(图2d)。上述结果表明,当LiTFSI与MoS2相互作用时,C1-S1键倾向于断裂。显然,DFT结果与在CF3SO2-基团中产生缺电子S中心的假设机制非常吻合。
为了确定缺电子S原子与DOL溶剂之间的相互作用,计算了去除LiCF3后DOL分子与LiTFSI剩余部分(即CF3SO2N–S+O2残基,图2e)之间的相互作用。键长分析显示,C2-O3和C3-O3键分别增加了0.05和0.03埃,而C2-O4键长缩短了0.03埃(图2f)。因此,由于与LiTFSI残基的不对称相互作用和C2-O3键断裂倾向于断裂,原本对称的DOL分子变得扭曲,并诱导了进一步的DOL聚合。上述计算结果进一步证明了图2b中假设机制的有效性,表明DOL聚合是由缺电子S中心触发的。
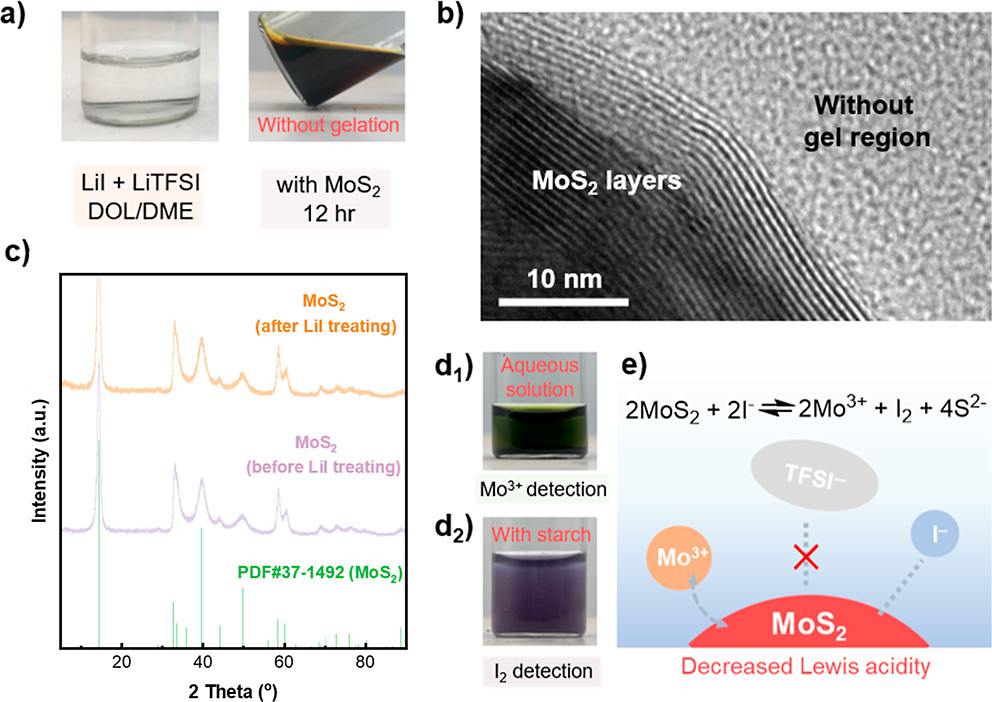
【图3】(a)LiI的凝胶抑制现象的光学图像。(b)用LiI溶液处理后的MoS2电催化剂HRTEM图像。(c)用LiI溶液处理前后MoS2电催化剂的XRD图谱。添加MoS2后检测LiI溶液中的(d1)Mo3+和(d2)I2。(e)LiI、MoS2和LiTFSI之间相互作用的示意图。
基于上述分析,削弱MoS2和LiTFSI之间的相互作用以避免缺电子路易斯酸性S中心的产生,理论上可以有效抑制MoS2电催化剂的表面凝胶化并促进其电催化性能。考虑到LiI的Lewis碱性,将LiI作为添加剂引入电解质中以阻断MoS2和LiTFSI之间的相互作用并抑制表面凝胶化。图3a显示,将溶解在DOL/1,2-二甲氧基乙烷(DME)中的1.0mol L-1LiTFSI中作为对照,而在空白溶液中额外加入0.10mol L-1LiI,表示为LiI溶液。在上述两种溶液中加入MoS2颗粒后,空白溶液在10min内变成准固相,而LiI溶液在12h后仍保持流动性。高分辨率TEM(HRTEM)图像显示,MoS2层周围没有凝胶区域(图3b),表明MoS2的表面在浸入LiI溶液后保持干净,有效地抑制了表面凝胶化。上述结果表明,路易斯碱性LiI确实阻断了LiTFSI和MoS2之间的相互作用,从而抑制了凝胶化发生。
添加MoS2颗粒后LiI溶液的颜色变为深棕色,表明在凝胶抑制过程中,LiI和MoS2之间发生了强烈反应。在LiI处理前后,MoS2的XRD图谱中没有观察到明显的峰位置变化,表明MoS2的整体结构没有显着变化(图3c)。通过直接观察溶解物质的特征颜色来检测电解质中的物质变化(图3d)。深绿色代表Mo3+的特征颜色,添加淀粉后LiI溶液出现紫色,表明I2的生成。因此,LiI和MoS2之间的反应如下
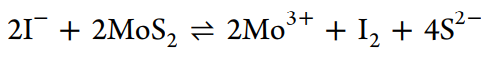
因此,LiI有效抑制了MoS2电催化剂上的表面凝胶化(图3e)。LiI与MoS2的反应削弱了LiTFSI与MoS2之间的相互作用,阻止了缺电子S中心的产生,避免了凝胶化加剧现象。此外,Lewis酸性MoS2被Lewis碱性I-包围,部分还原为具有较低Lewis酸度的Mo3+,从而降低了MoS2电催化剂的整体Lewis酸度并消除了表面凝胶化。
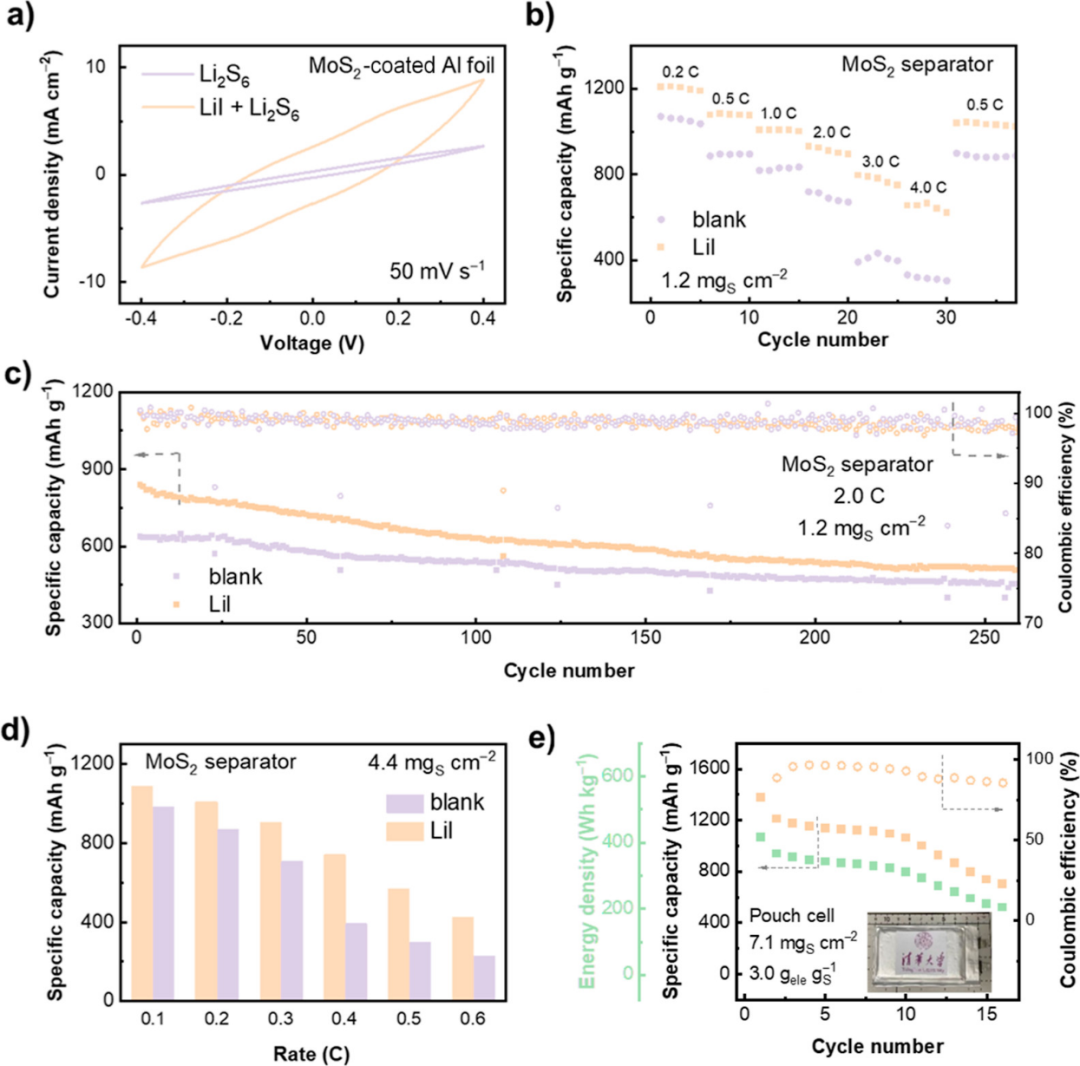
【图4】(a)有或没有LiI的Li2S6对称电池CV曲线。(b)锂硫纽扣电池的倍率性能和(c)2.0C下的长循环性能。(d)具有高硫载量的Li-S纽扣电池倍率性能。(e)400Wh kg–1级软包电池的循环性能。
图4a显示,在CV曲线中,与没有LiI的Li2S6对称电池相比,具有LiI的Li2S6对称电池表现出2.3倍的峰值电流密度,表明液-液氧化还原动力学得到改善。图4b显示,在常规硫载量(1.2mgS cm-2)下,具有LiI的电池在0.2、0.5、1.0、2.0、3.0和4.0C(1C=1672 mA gS-1)下表现出优异的放电比容量,分别为1208、1077、1006、931、795和655mAh g-1。图4c显示,即使在2.0C的高循环倍率下,含LiI的Li-S纽扣电池初始放电比容量也比不含LiI的电池高200mAh g-1,并且可稳定循环超过250次。
在实际条件下进一步组装和评估了具有4.4mgScm-2高硫载量的Li-S纽扣电池。图4d显示,在0.4C下,含LiI的电池具有742mAh g-1的高放电比容量,而不含LiI的电池仅获得了393mAhg-1的放电比容量。为了充分证明在实际高能量密度器件中使用电催化MoS2和LiI抑制凝胶的可行性,组装了具有MoS2电催化剂和LiI电解质的2.5Ah级Li-S软包电池。硫负载量控制在7.1mgScm-2,对于400Wh kg-1级Li-S软包电池,电解液与硫(E/S)比设置为3.0g电解液gS-1。400Wh kg-1级Li-S软包电池实际能量密度为416Wh kg-1,初始比容量为1380mAh gS-1,可稳定循环16个循环,并保持较高是库仑效率(图4e)。
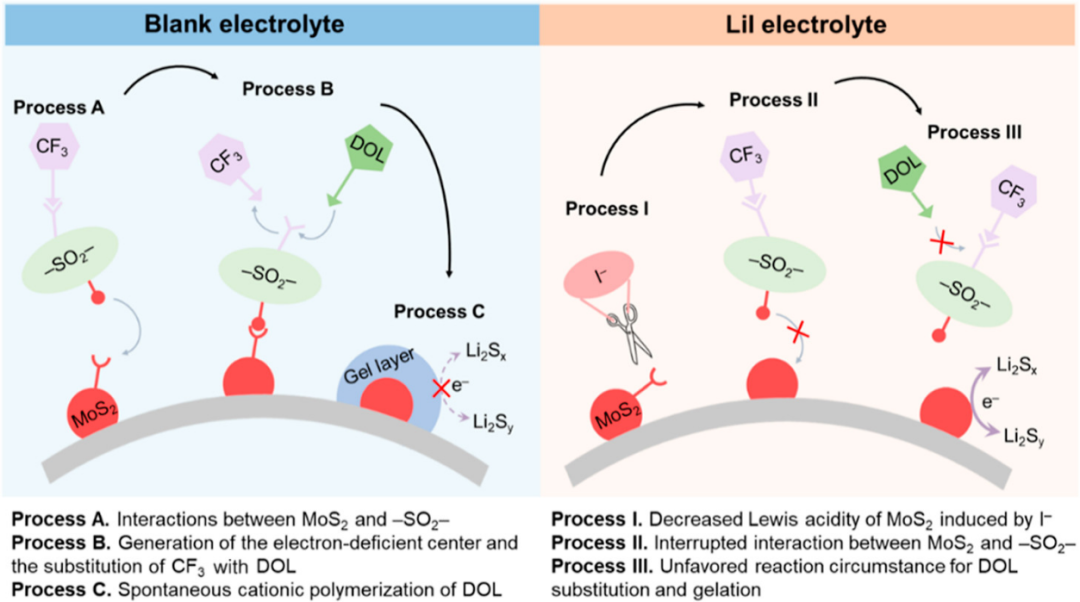
【图5】Li-S电池中空白电解质(左)和LiI电解质(右)中的反应路径示意图。
锂盐调控DOL在MoS2上表面凝胶化的机理及相应影响如图5所示。在空白电解液中,LiTFSI中的CF3SO2-基团与MoS2上的Lewis酸性位点相互作用,导致CF3-损失,并产生缺电子中心。与原始MoS2相比,缺电子中心表现出更高的路易斯酸度,与DOL溶剂强烈相互作用,加剧了MoS2电催化剂上的表面凝胶化,并降低了硫氧化还原反应的电催化活性(图5)。然而,在LiI电解质中,Lewis碱性I-优先与MoS2上的Lewis酸性位点相互作用,并阻止MoS2和LiTFSI之间的相互作用。因此,DOL的聚合受到抑制,MoS2的电催化活性恢复,从而提高了电池性能(图5)。
04总结和展望
Li-S电池中MoS2电催化剂、DOL溶剂和LiTFSI盐之间的相互作用表明,LiTFSI加剧了DOL在MoS2电催化剂上的表面凝胶化。LiTFSI中的CF3SO2-基团与MoS2电催化剂上的路易斯酸性位点相互作用,产生一个缺电子中心,加剧了DOL的阳离子聚合。凝胶层覆盖了MoS2电催化剂的表面并降低了电催化活性。为了解决上述问题,本工作将路易斯碱性添加剂LiI引入Li-S电池中,以阻断MoS2与LiTFSI之间的相互作用,避免产生缺电子中心,抑制表面凝胶化。因此,具有LiI添加剂和MoS2电催化剂的Li-S电池表现出更优异的倍率性能、更高的放电容量(1380mAh gS-1)和416Whkg–1的超高实际能量密度。这项工作提供了一种有效的锂盐来提高实际工作中锂硫电池的电催化活性,并加深了对储能系统中电催化剂、溶剂和盐之间相互作用的基本理解。
参考文献:
Xi-Yao Li, Shuai Feng, Chang-XinZhao, Qian Cheng, Zi-Xian Chen, Shu-Yu Sun, Xiang Chen, Xue-QiangZhang, Bo-Quan Li*, Jia-Qi Huang, and Qiang Zhang*. RegulatingLithium Salt to Inhibit Surface Gelation on an Electrocatalyst forHigh-Energy-Density Lithium–Sulfur Batteries, Journalof the American Chemical Society.
DOI:10.1021/jacs.2c04176
https://doi.org/10.1021/jacs.2c04176
转自:清新电源

